

Translate this page into:
Rare Cause of Secondary Pulmonary Arterial Hypertension
Anand Yadav Pasula, MBBS, MD, DM Department of Cardiology, Nizam's Institute of Medical Sciences Hyderabad 500082, Telangana India anand6pasula@gmail.com
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Unilateral absent pulmonary artery (UAPA) is a rare congenital disorder. Most of the patients will be diagnosed in the early childhood because of recurrent respiratory tract infections and hemoptysis, but adult presentation is not uncommon. We report a 47-year-old female who was earlier diagnosed as bronchiectasis with pulmonary artery hypertension but now presented with complaints of typical angina and dyspnea on exertion. During right heart catheterization we failed to enter right pulmonary artery (RPA), and conventional coronary angiogram showed a large left atrial branch of left circumflex giving collateral blood supply to the right lung. Computed tomography angiogram showed absent RPA. We report this case not only because the patient was misdiagnosed but also because of rarity of coronary collaterals in UAPA patients and unusual bilateral bronchiectasis. According to reported literature, ours is the 28th case of this nature.
Keywords
pulmonary artery hypertension
absent right pulmonary artery
coronary collateral

Introduction
Unilateral absent pulmonary artery (UAPA) is a rare congenital anomaly with incidence of 1 in 200,000 individuals. It is usually associated with cardiovascular anomalies like tetralogy of Fallot, atrial septal defect, truncus arteriosus, coarctation of aorta, right aortic arch, patent ductus arteriosus, and supravalvular aortic stenosis. Majority of the patients will be diagnosed early in life, but delayed presentations have been reported in the literature. In most of the patients, systemic collaterals will supply the affected lung, and only in few patients coronary collaterals were observed.
Case Report
A 47-year-old female previously diagnosed as bilateral bronchiectasis was now admitted for worsening of breathlessness with pedal edema and chest pain for the last 1 month. No past history of tuberculosis or pertussis and recurrent respiratory tract infections in the childhood was found. She is mother of three children and pregnancies were uneventful and all her children are healthy. On examination, patient was having cyanosis with body mass index of 21.1 kg/m2; her vitals were stable except decreased oxygen saturation on room air, 82%; with 2 L of supplementation oxygen saturation was improved to 94%, suggesting pulmonary pathology as the cause for cyanosis. Cardiovascular examination revealed palpable P2, grade 2 parasternal heave, Loud P2, and no murmurs. Coarse crepitations were noted in the bilateral lower lung fields.
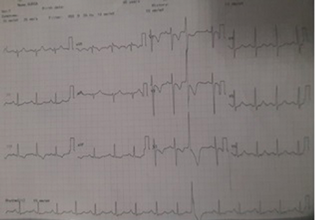
Electrocardiogram (ECG) showed right axis deviation with mean frontal QRS Complex axis of 120 and T-wave inversions in leads V1-V3, III, and Augmented vector foot (aVF) with occasional ventricular premature complexes (Fig. 1, ECG image). Echocardiography showed dilated right atrium and right ventricle with severe tricuspid regurgitation and severe pulmonary arterial hypertension (PAH). Pulmonary artery branching pattern was not appreciated in the echocardiography because of mediastinal shift and poor echo window. Chest radiograph showed reduced pulmonary vascularity and loss of lung volume on right side with compensatory hyperinflated left lung and shift of mediastinal structures to right side and bronchiectatic features in both lung fields. Pulmonary function test showed severe restrictive pattern and no obstructive component with poor bronchodilator response. Pulmonary function test FEV1/FVC—forced expiratory volume in one second/forced vital capacity—is 75.3 (FEV1 26%) and postbronchodilator FEV1 27%.
-
Fig. 1 Electrocardiogram of the patient.
Fig. 1 Electrocardiogram of the patient.
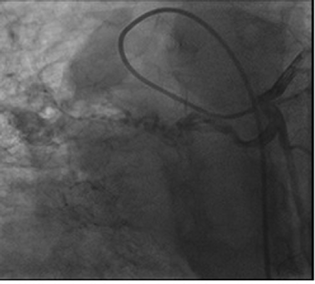
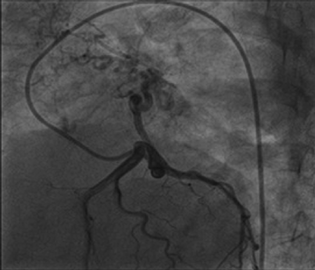
Right heart catheterization showed baseline and postoxygenation mean pulmonary artery pressure of 66 mm Hg and 35 mm Hg, respectively. Pulmonary capillary wedge was 3 mm Hg. During the procedure, Swan Ganz catheter failed to enter in right pulmonary artery (RPA). Calculated cardiac output was 7.9 l/min. Conventional Coronary angiogram revealed large left atrial branch from left circumflex artery (LCX), which is aneurysmally dilated and arborizing in right lower lobe of lung, without any coronary artery obstruction (Figs. 2 and 3). Angina may be due to steal of blood from the LCX to lung parenchyma.
-
Fig. 2 Aneurysmal collateral branch from the left circumflex artery.
Fig. 2 Aneurysmal collateral branch from the left circumflex artery.
-
Fig. 3 Left circumflex artery provides blood supply to right lung.
Fig. 3 Left circumflex artery provides blood supply to right lung.
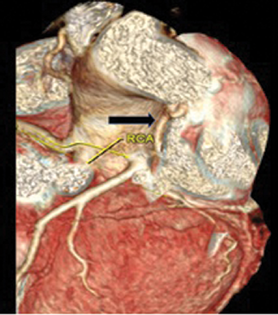
High-resolution computed tomography (HRCT) chest showed bilateral bronchiectasis with bronchial wall thickening noted in the right upper lobe, right lower lobe, and lingular segment of left upper lobe (Fig. 4). Cystic bronchiectatic changes were noted in the contralateral lower lobe of left lung, which is rare finding in patients with UAPA. Interstitial thickening was noted in right lower lobe, which mimics localized pulmonary edema. Other classical features of UAPA like small ipsilateral hemithorax, elevated diaphragm, and mediastinal shift were also present. Computed tomography (CT) angiogram showed absent RPA (Fig. 5) with evidence of other multiple systemic collaterals from right subclavian artery, few collaterals arising directly from under surface of aortic arch, and one collateral from celiac trunk ascending into right hemithorax through diaphragmatic foramen and coronary collateral from left circumflex supplying right lung (Figs. 6 and 7, images of coronary collateral). CT venogram showed blood from right lung draining abnormally into prominently dilated azygous vein (9 mm diameter).

-
Fig. 4 High-resolution computed tomography chest showing bronchiectasis in the middle lobe and lingular segment of the left upper lobe.
Fig. 4 High-resolution computed tomography chest showing bronchiectasis in the middle lobe and lingular segment of the left upper lobe.

-
Fig. 5 Computed tomography pulmonary angiogram showing absent right pulmonary artery.
Fig. 5 Computed tomography pulmonary angiogram showing absent right pulmonary artery.
-
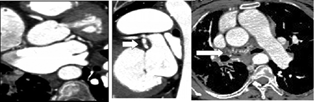
Fig. 6 Volume-rendered image depicting dilated left atrial branch (black arrow).
Fig. 6 Volume-rendered image depicting dilated left atrial branch (black arrow).
-
Fig. 7 Computed tomography coronary angiogram reveals dilated left atrial branch (arrow) from the left circumflex artery distally continuing as multiple fistulae into lung parenchyma (arrow).
Fig. 7 Computed tomography coronary angiogram reveals dilated left atrial branch (arrow) from the left circumflex artery distally continuing as multiple fistulae into lung parenchyma (arrow).
Patient was treated in cardiology intensive care unit with diuretics, vasodilators such as calcium channel blocker, and Tadalafil with ambrisentan and continuous low flow oxygen. Patient improved and was discharged with advice to take home oxygen therapy, vaccinations, and antipulmonary artery hypertension drugs.
Discussion
UAPA is a rare congenital cardiovascular abnormality with an estimated prevalence of 1 in 200,000 individuals.1 Abnormal involution of sixth aortic arch results in branch pulmonary artery agenesis.2 In most of the patients, distal part of intrapulmonary arterial system remains patent with collateral blood supply from aortic branches and rarely from coronary arteries. Among UAPA patients, RPA is involved two times more commonly than left pulmonary artery (LPA). Associated cardiac anomalies are more frequent with absent LPA rather than RPA. Most cases of absent RPA are usually present as isolated congenital anomaly.
Ipsilateral bronchiectasis of the affected lung is more common in UAPA. In our patient, bilateral bronchiectasis with severe restrictive pattern of pulmonary function test is noticed. Bilateral bronchiectasis is rare in UAPA patients, as till now only two cases were reported. Pathogenesis of bronchiectasis is probably due to poor perfusion of lung with decreased inflammatory cell migration, impaired ciliary motility, and mucus entrapment followed by secondary infection and bronchiectasis. Our patient had severe PAH, which could be due to two reasons: one of them is increased pulmonary blood flow to LPA leading to increased shear stress on vascular endothelium and increased endothelin production, and second reason is ventilation and perfusion mismatch causing hypoxia-induced vasoconstriction and PAH. In our patient, we observed reversible component of PAH on right heart catheterization study with oxygen supplementation.
Darwazah and Alhaddad reported 2 cases and also reviewed 13 UAPA patients with coronary collaterals in whom 54% had associated systemic collaterals similar to our patient, whereas rest of them had only coronary collaterals. Most of the times coronary collaterals are from LCX (61%) followed by right coronary artery (36%) and left anterior descending artery (3%).3 Later, Baştuğ et al reported a case of a 71-year-old woman with absent LPA and collateralization from all three major coronary arteries.4 According to reported literature, till now only 27 cases of UAPA with coronary collaterals were published and in addition to that our patient has a rare feature of bilateral bronchiectasis.
High index of suspicion is required to diagnose UAPA, otherwise delay or misdiagnosis can occur. Features that should alert for possibility of UAPA are chest radiograph showing unilateral decrease in pulmonary vascularity, echocardiographic failure to delineate branching pattern of pulmonary artery and failure to enter into one of the branches of main pulmonary artery during right heart catheterization, and HRCT chest showing features of unilateral pulmonary edema, unilateral bronchiectasis, and unilateral loss of lung volume with shift of mediastinal structures.5 CT pulmonary angiogram is the investigation of choice to diagnose UAPA. Coronary angiogram and aortogram will give information of major aortopulmonary collateral arteries to the affected lung.
There are no standard guidelines on the treatment of UAPA. Multiple surgical revascularization techniques have been tried such as end-to-end direct anastomosis and reconstruction of neo-pulmonary artery using ligamentum arteriosus, revascularization using saphenous vein graft, and prosthetic conduit. Patients with recurrent pulmonary infections and massive hemoptysis can be treated with lobectomy or pneumonectomy and selective embolization of systemic collaterals, respectively.6 Patients in whom revascularization is difficult and with moderate-to-severe PAH causing symptoms can be managed with antipulmonary artery hypertension drugs as in our case.7
Conclusion
Delayed presentation of UAPA in the middle age group is not uncommon. Coexistent lung pathology can mislead and delay its diagnosis. Hence, it is imperative to evaluate pulmonary vascular abnormality in patients having bronchiectasis with unusual cyanosis and disproportionate PAH.
Conflict of Interest
None declared.
References
- The varied manifestation of pulmonary artery agenesis in adulthood. Chest. 1995;108(03):670-676.
- [Google Scholar]
- Absence of a primary division of the pulmonary trunk. An ontogenetic theory. Circulation. 1964;29:124-131.
- [Google Scholar]
- Pulmonary artery agenesis associated with coronary collaterals among adults. J Cardiothorac Surg. 2016;11(01):109.
- [Google Scholar]
- Congenital absence of left pulmonary artery with collateralization from all major coronary arteries. Turk Kardiyol Dern Ars. 2016;44(03):240-243.
- [Google Scholar]
- Unilateral proximal interruption of the pulmonary artery in adults: CT findings in eight patients. J Comput Assist Tomogr. 2002;26(05):777-783.
- [Google Scholar]
- Congenital isolated unilateral agenesis of pulmonary arteries in adults: case series and review. Indian J Thorac Cardiovasc Surg. 2021;37(01):144-154.
- [Google Scholar]
- Idiopathic Pulmonary Artery Hypertension. May 7. In: StatPearls [Internet]. Treasure Island, FL: StatPearls Publishing; 2021 Jan-. PMID: 29489262
- [Google Scholar]




