

Translate this page into:
Pulmonary Hypertension: Diagnosis and Management

*Corresponding author: Sridevi Chigullapalli, Department of Cardiology, Dr. D. Y. Patil Medical College and Hospital, DY Patil Vidyapeeth, Pimpri-Chinchwad, Maharashtra, India. csridevi2007@rediffmail.com
-
Received: ,
Accepted: ,
How to cite this article: Chigullapalli S, Malani SK. Pulmonary Hypertension: Diagnosis and Management. Indian J Cardiovasc Dis Women. 2024;9:167-75. doi: 10.25259/IJCDW_35_2024
Abstract
Pulmonary hypertension (PH) affects 1% of people worldwide. Changes in the pulmonary vasculature, obstructive lesions in the pulmonary arteries, and an increase in pulmonary artery pressure are the hallmarks of PH, a progressive and deadly cardiovascular illness. These modifications result in a rise in right ventricular afterload, which frequently causes unfavorable right ventricular remodeling, right ventricular dysfunction and, in the end, mortality. One of the more severe and well-researched types of PH is pulmonary arterial hypertension (PAH), which is treatable with medication. The mechanisms involved in the regulation of pulmonary vascular tone and proliferation are the focus of PAH as well as some other forms of PH. The main characteristics of PAH (group 1) are discussed in this article, along with new and existing treatment options for the condition.
Keywords
Pulmonary hypertension
Classification
2022 European Society of Cardiology/European Respiratory Society guidelines

INTRODUCTION
Pulmonary arterial hypertension (PAH) previously known as primary pulmonary hypertension (PH) was defined as a mean pulmonary artery pressure (mPAP) of >25 mmHg at rest during the First World Symposium on PH, which took place in Geneva in 1973.[1] The Sixth World Symposium on PH which was held in 2018 changed the definition of PAH to mPAP more than 20 mmHg and included pulmonary vascular resistance (PVR) 3 Woods units or more.[2]
CLASSIFICATION OF PH
PH is classified into 5 groups based on hemodynamics and associated other systemic diseases.
Table 1 illustrates the five clinical classes into which PH is classified and hemodynamics in various classes of hypertension and treatment of PH.
| Classification | Hemodynamics | Treatment |
|---|---|---|
| Group 1 | ||
|
Pre-capillary pulmonary hypertension Mean PA pressure >20 mmHg, PVR ≥3 Wood units, PAWP ≤15 mmHg |
Pulmonary vasodilators |
| Group 2 | ||
|
Post-capillary pulmonary hypertension Mean PA pressure >20 mmHg, PVR ≤3 Wood units, PAWP ≥15 mmHg. |
Treat left heart disease |
| Group 3 | ||
|
Pre-capillary pulmonary hypertension Mean PA pressure >20 mmHg, PVR ≥3 Wood units, PAWP ≤15 mmHg. |
Treat underlying lung disease Supplemental oxygen Inhaled treprostinil approved for ILD PH only |
| Group 4 | ||
|
Pre-capillary pulmonary hypertension Mean PA pressure >20 mmHg, PVR ≥3 Wood units, PAWP ≤15 mmHg. |
Treat underlying disease Chronic thromboembolism is considered thromboendarterectomy. Balloon angioplasty and soluble guanylyl cyclase inhibitors |
| Group 5. Miscellaneous | ||
|
Predominantly pre-capillary but post-capillary and combined pre-capillary and post-capillary are also included | Treat underlying cause Off-label use of pulmonary vasodilators in selected patients |
PAH: Pulmonary arterial hypertension, PVR: Pulmonary vascular resistance, HIV: Human immunodeficiency virus, PAWP: Pulmonary artery wedge pressure, PH: Pulmonary hypertension, ILD: ILD: Interstitial lung disease, PPH: Persistent Pulmonary Hypertension, COPD: Chronic obstructive
CLINICAL ASSESSMENT AND INVESTIGATIONS
To correctly assign a clinical group and identify suspected cases of PH, a thorough clinical assessment is necessary. Using history alone, it is difficult to distinguish PAH’s clinical symptoms from those of other cardiorespiratory disorders because they are frequently non-specific. Dyspnea, exhaustion, syncope, and symptoms of right heart failure are common in PAH. It is important to obtain information on past medical history, diet, medication and drug exposures, and family history of PAH. Signs of the underlying etiology may be seen on a clinical examination. The presence of central cyanosis and clubbing of fingers and toes suggest the presence of cyanotic congenital heart disease, whereas the presence of sclerodactyly, digital ulcers, and telangiectasia suggests systemic sclerosis as a cause of PAH. Signs of right heart failure should be looked for which will help in risk stratification and to start treatment with diuretic therapy.
HAEMATOLOGICAL INVESTIGATIONS
Routine blood investigations such as complete blood picture, liver function test, renal function test, thyroid function tests, brain natriuretic peptide (BNP), and N-terminal pro-BNP (NT-proBNP) testing are done in all patients. A number of additional blood tests that should be carried out at baseline for conditions associated with PAH are highlighted in Table 1. These include human immunodeficiency virus (HIV) and hepatitis screenings, as well as panels for connective tissue diseases, such as serum antinuclear antibodies, anti-centromere antibodies, and ds DNA antibodies.
PULMONARY FUNCTION TEST
In PH, pulmonary function tests (PFTs), give useful information. Spirometry is a useful tool for evaluating the presence of underlying restructive or obstructive lung diseases, and lung carbon monoxide diffusion capacity (DLCO) can yield prognostic data.[3] DLCO is reduced in patients with restrictive lung disease and pulmonary veno occlusive disease (PVOD). These tests are helpful in classifying PH patients into different subgroups. Specific subgroups of PAH, including idiopathic PAH, suspected hereditary PAH, and PVOD, should be diagnosed based on genetic counseling and investigations to rule out secondary causes.[4]
PATHOPHYSIOLOGY OF PAH
Characteristic features of pulmonary circulation are low resistance system and pressure is one-tenth of systemic pressure. Pulmonary vasculopathy of PAH is characterized by vasoconstriction, vascular remodeling of three layers of the artery wall, and in situ thrombosis [Supplementary Figure 1]. Patients with plexiform lesions, vascular abnormalities marked by complex vascular formations emanating from remodeled pulmonary arteries, are seen in the most advanced stage of the disease.[5-8] These changes cause gradual luminal constriction, elevated PVR, and right ventricular afterload, which ultimately leads to right heart failure.
An imbalance of vasoactive mediators, including an excess of endothelin and thromboxane and a relative lack of prostacyclin and nitric oxide, is what defines PAH[9] are responsible for the above pathophysiological changes. Prostacyclin and nitric oxide are significant vasodilators in the pulmonary circulation, and replacement is a key component of treatment.[10] On the other hand, PAH causes a rise in endothelin -1, therefore blocking this pathway is another important part of the treatment strategy. In PAH, there is an imbalance in the signaling of transforming growth factor-beta (TGF-β) and bone morphogenetic protein (BMP), with less signaling through the BMP route and more signaling through the TGF-β pathway[11] and sotatercept is a new drug which targets this pathway.
RADIOLOGICAL INVESTIGATIONS
Ventilation–perfusion (V/Q) imaging, computed tomography (CT) of the thorax, and chest radiography are useful to rule out pulmonary causes for PH. A CT pulmonary angiogram can be used to evaluate the presence of chronic thromboembolism PH (CTEPH) and parenchymal lung disease. Lung V’/Q’ imaging is crucial to rule out distal CTEPH. To rule out cirrhosis and underlying portal hypertension, a Doppler ultrasound of the liver and portal system should be carried out. Cardiac magnetic resonance imaging (MRI) is included in the updated risk stratification table in 2022 European Society of Cardiology/European Respiratory Society (ESC/ ERS) guidelines for diagnosis and assessment of PH.[12] Various MRI parameters useful for risk stratification are stroke volume index, right ventricular ejection fraction, and right ventricular end-systolic volume index.
ECHOCARDIOGRAPHY
One of the most useful diagnostic tests for people with suspected PH is echocardiography. The probability of PH can be estimated in symptomatic subjects by measuring the peak tricuspid regurgitation jet velocity (TRV) and other echocardiographic variables.[5] The probability of PAH can be categorized as low, intermediate, or high, which can help determine which patients would benefit from further right heart catheterization (RHC). A peak TRV >3.4 m•s−1 in a patient exhibiting symptoms indicates a high likelihood of PH. On the other hand, a peak TRV of <2.8 m/s with no additional echocardiography features and no symptoms suggests low likelihood of PH.[12] In subjects with established PAH, echocardiographic parameters also make comprehensive risk assessment easier.[2] Prognostic parameters in PAH are right atrial area, the presence of a pericardial effusion, and the ratio of tricuspid annular plane systolic excursion (TAPSE) to pulmonary arterial systolic pressure (PASP). The TAPSE/PASP ratio is a reliable noninvasive indicator of right ventricle and pulmonary artery coupling.
RHC
RHC is helpful in assessing pulmonary hemodynamics. Dr. Werner Forssmann conducted the first human RHC on himself in 1929.[13] Cournand and Richards improved this later in the 1940s, and their joint effort was awarded the Nobel Prize in 1956. RHC and vasoreactivity tests should be done in advanced centers to prevent the serious side effects associated with these tests.[14]
Patients are categorized into one of five clinical groups based on clinical features and findings from RHC. A resting mPAP at RHC of more than 20 mmHg indicates PH.[1] The pulmonary artery wedge pressure (PAWP), PVR, and mPAP can all be used to characterize the hemodynamic pattern after PH has been established.[15]
Hemodynamic patterns of PH
Pre-capillary: m PAP >20 mmHg, PVR ≥3Wood units, PAWP ≤15 mmHg
Post-capillary: m PAP >20 mmHg, PVR ≤3 Wood units, PAWP ≥15 mmHg
Combined: m PAP >20 mmHg, PVR ≥3 Wood units, PAWP ≥15 mmHg.
Vasoreactivity testing
All patients with idiopathic PAH, hereditary PAH, or drug-induced PAH should undergo acute vasoreactivity testing. This is performed by giving a vasoactive agent – such as iloprost or nitric oxide – by inhalation during right heart catheterization.[15]
During vasoreactivity testing, <10% of patients will show an acute response, and even fewer will have long-term responsiveness, those who do, however, usually have a good prognosis and can be treated with long-term calcium channel blockers (CCBs).[16]
Vasoreactivity testing is not commonly used in other PAH subgroups due to its low diagnostic yield and potential for unfavorable side effects, including pulmonary edema.
The following are the definitions of acute and long-term responder.[16]
Acute responder
m PAP reduction of 10 mmHg or more or to a value of 40 mmHg or less
Cardiac output increased or maintained but not reduced.
Long-time responder
Persistent clinical, biochemical, and hemodynamic improvement to CCBs for more than 12 months.
RISK STRATIFICATION
In PAH, comprehensive risk assessment is a complex, dynamic, and important part of individualized patient care. For risk stratification, numerous scores, tables, and equations have been created. The European Society of Cardiology/European Respiratory Society (ESC/ERS) risk stratification table and the risk equations and scores from the Registry to Evaluate Early and Long-term PAH Disease Management (REVEAL) registry are two important risk stratification tools.[17-19] The updated 2022 guidelines[12] feature an updated ESC/ERS risk stratification table using multiple clinical and investigations [Table 2]. These variables are signs of right heart failure, progression of disease symptoms and clinical manifestations, syncope, World Health Organization functional class, 6-min walking distance (6MWD), cardiopulmonary exercise test, BNP and NT-ProBNP levels, echocardiography and cardiac MRI, and hemodynamics [Table 3].
| Determinants of prognosis (estimated 1-year mortality) | Low risk (<5%) | Intermediate risk (5–20%) | High risk ( >20%) |
|---|---|---|---|
| Signs of right heart failure | Absent | Absent | Present |
| Progression of disease symptoms and clinical manifestation | No | Slow | Rapid |
| Syncope | No | Occasional | Repeated |
| WHO-FC | I and II | III | IV |
| 6MWD | >440 | 165–440m | <165 m |
| CPET | Peak VO2>65% predicted | Peak VO2 35–65% predicted | Peak VO2<35% predicted |
| BNP and NT-ProBNP (ng/L) | • BNP <50 • NT-proBNP<300 |
• BNP 50–800 • NT –proBNP 300–1, 100 |
BNP >800 NT-pro BNP >1, 100 |
| Echocardiography | • RA area<18 cm2 • TAPSE/s PAP >0.32 • No pericardial effusion |
• RA area 18–26 cm2 • TAPSE/s PAP 0.19–0.32 • Minimal pericardial effusion |
• RA area<18 cm2 • TAPSE/s PAP <0.19 • Moderate-to-large pericardial effusion |
| Cardiac MRI | • RVEF >54% • SVI >40 mL/m2 • RVESVI<42 mL/m2 |
• RVEF 37–54% • SVI 26–40 mL/m2 • RVESVI 42–54 mL/m2 |
• RVEF <37% • SVI <26 mL/m2 • RVESVI >54 mL/m2 |
| Hemodynamics | • RAP<8 mmHg • CI ≥2.5 • SVI >38 mL/m2 • SvO2>65% |
• RAP 8-14 mmHg • CI=2–2.4 • SVI=31–38 mL/m2 • SvO2=60–65% |
• RAP <8 mmHg • CI <2 • SVI <31 mL/m2 • SvO2<60% |
ESC/ERS: European society of cardiology/European respiratory society, PAH: Pulmonary arterial hypertension, WHO-FC: World Health Organization functional class, 6MWD: 6-min walking distance, BNP: Brain natriuretic peptide, CI: Confidence interval, CPET: Cardiopulmonary exercise testing, MRI: Magnetic resonance imaging, NT–proBNP: N-terminal prohormone of brain natriuretic peptide, RAP: Right atrial pressure, TAPSE: Tricuspid annular plane systolic excursion, RVEF: Right ventricular ejection fraction, SVI: Systolic volume index, RVESVI: Right ventricular end-systolic volume index, PAP: Pulmonary arterial pressure, SvO2: Venous oxygen saturation, VO2: Volume of oxygen
| Determinants of prognosis | Low risk | Intermediate- low risk | Intermediate- high risk | High risk |
|---|---|---|---|---|
| Points assigned | 1 | 2 | 3 | 4 |
| WHO-FC | I or II | - | III | IV |
| 6MWD | >440 | 320–440 | 165–319 | <165 |
| BNP or | <50 | 50–199 | 200–800 | <800 |
| NT-proBNP, ng/L | <300 | 300–649 | 650–1100 | <1100 |
6 MWD: 6 minute walking distance, BNP: Brain natriuretic peptide, NT-proBNP: N-terminal pro-brain natriuretic peptide, WHO-FC: World Health Organization functional class. Risk is calculated by dividing the sum of all grades by the number of variables of variables and rounding to the next integer. WHO-FC I and II are assigned 1 point as both are associated with good long-term survival
The 1-year mortality risk is estimated using multiparametric risk assessment, which also stratifies patients into low-risk (5%), intermediate-risk (5–20%), and high-risk (20%) groups. A non-invasive four-stratum model utilizing NT-proBNP, 6MWD, and World Health Organization functional class is now advised for follow-up visits. Patients are categorized as high-risk, low-risk, intermediate-high-risk, and low-risk.
TREATMENT OF PULMONARY ARTERY HYPERTENSION
A person-centered, dynamic approach is needed because of the progressive nature of the disease. There is rarely a “one-size-fits-all” solution, and each patient’s needs and characteristics are unique and subject to change over time. An overview of PAH-specific therapy and crucial supportive measures are given in this section. Table 4 highlights the do”s and dont's from recent ESC/ERS 2022 guidelines.
| Recommended | Not recommended |
|---|---|
| RHC to establish diagnosis of PAH/CTEPH or in diagnostic dilemma Echocardiography as first-line test in suspected PH. TRV >2.8 m/s should be taken as the probability threshold of PH Risk stratification at the time of diagnosis and during follow-ups using three strata model Supervised exercise training after clinical stabilization Lifelong therapeutic anticoagulation patients with CTEPH VKAs for patients with APLA Initial combination therapy with ambrisentan and tadalafil or macitentan and tadalafil In patients with ASD, VSD, or PDA and a PVR <3 WU, go for shunt closure Bosentan in symptomatic Eisenmenger syndrome Inhaled treprostinil in patients with PH associated with ILD irrespective of PH - severity |
Vasoreactivity testing in patients with PAH other than I/H/D PAH and in PH groups 2,3,4, and 5 Use of ACEis, ARBs, ARNI, SGLT-2ls, beta-blockers, or ivabradine in PAH unless required by comorbidities Calcium channel blockers without vasoreactivity testing Initial combination therapy with macitentan, tadalafil, and selexipag Addition of riociguat to PDE-5 is Pregnancy is contraindicated in PH Combining bosentan and sildenafil to reduce morbidity/mortality In patients with ASD and a PVR >5 WU, despite PAH treatment, shunt closure is contraindicated Use of PAH medications in unclassified PH Use of PAH medications in patients with lung disease with non-severe PH |
ACEis: Angiotensin-converting enzyme inhibitors, APLA: Antiphospholipid antibody, ARBs: Angiotensin receptor blockers, ARNI: Angiotensin receptor neprilysin inhibitor, ASD: Atrial septal defect, VSD: Ventricular septal defect, CTEPH: Chronic thromboembolism pulmonary hypertension, ESC/ERS: European society of cardiology/European respiratory society, I/H/D PAH: Idiopathic, heritable, drug-associated pulmonary arterial hypertension, ILD: Interstitial lung disease, PAH: Pulmonary arterial hypertension, PDA: Patent ductus arteriosus; PDE-5is: Phosphodiesterase-5 inhibitors, PH: Pulmonary hypertension, PVR: Pulmonary vascular resistance, SGLT-2ls: Sodium-glucose cotransporter-2-inhibitors, TRV: Tricuspid regurgitation velocity, VKAs: Vitamin K antagonists, RHC: Right heart catheterization, WU: Woods units
PAH-SPECIFIC THERAPY
For patients with PAH who respond well to high-dose CCBs exclusively over the long term, monotherapy is recommended. CCBs may be prescribed for different indications in other PAH subgroups, such as the Raynaud phenomenon in PAH associated with systemic sclerosis.
The three important treatment pathways for PAH are endothelin, nitric oxide, and prostacyclin are targeted for the treatment [Figure 1]. Reduced levels of endogenous nitric oxide and prostacyclin and increased levels of endothelin-1 are linked to endothelial dysfunction in the pulmonary vasculature in PAH [Figure 1]. Vasoactive mediators become unbalanced as a result, which causes vasoconstriction and remodeling of the pulmonary circulation. The goal of treatment is to bring this equilibrium back by inhibiting endothelin signaling and enhancing the nitric oxide and prostacyclin pathways. Majority of patients are advised to start initial double combination therapy (phosphodiesterase [PDE] inhibitors and endothelin receptor antagonists), as per the 2022 ESC/ERC guidelines [Figures 2 and 3] to attain low-risk profile. When a patient does not achieve low risk on double combination therapy, additional PAH-specific therapies (prostanoids or prostacyclin analogs) should be started. After starting PAH-specific therapy, patient should be reassessed after 1 month for risk assessment and risk stratification to escalate PAH therapy. These medication vasodilatory mechanisms have the potential to cause systemic side effects, especially when starting a new dosage and titrating it. Hypotension, flushing, dizziness, and gastrointestinal symptoms are common side effects after starting treatment. Other adverse effects include anemia (macitentan), redness of eyes, changes in color vision and photosensitivity (phosphodiesterase type-5 inhibitors like sildenafil) elevated aminotransferases (with bosentan), and low platelet count (parenteral prostacyclin). Individuals who are prescribed parenteral prostacyclin through central venous catheters need to take extra care because they have the risk of developing venous catheter infections

- Pathophysiology of pulmonary hypertension.
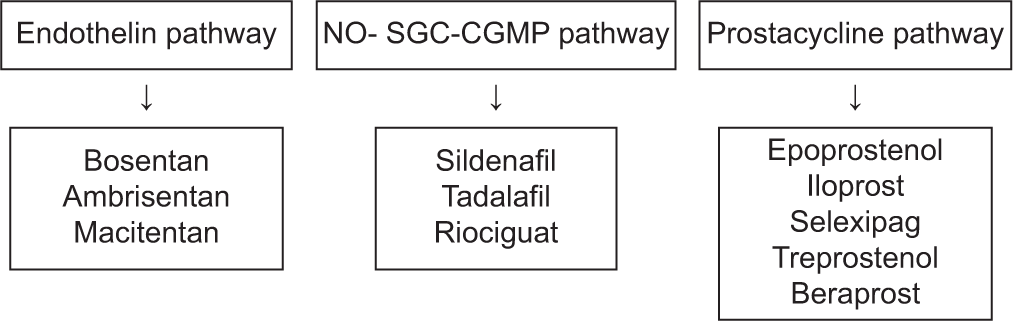
- Current therapeutic targets for pulmonary arterial hypertension. (NO-SGC-CGMP: Nitric oxide-Solble guanyl cyclase-Cyclic GMP)
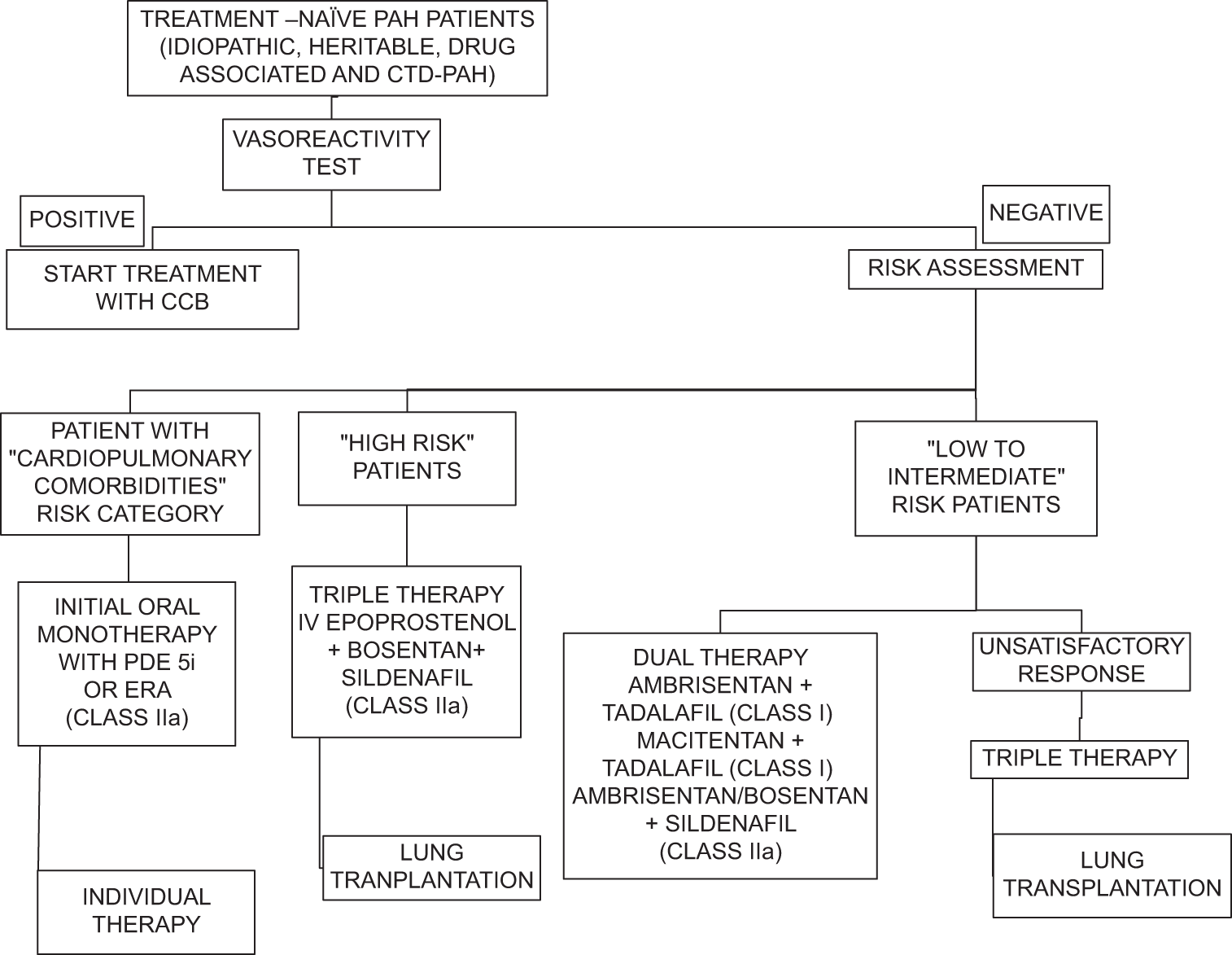
- Treatment algorithm for the management of pulmonary arterial hypertension as proposed by 2022 European society of cardiology/European respiratory society guidelines. (CTD-PAH: Connective tissue disease-Pulmonary arterial hypertension, CCB: Calcium channel blockers, PDE 5i: Pospho di esterase 5 inhibitors, ERA: Endothelin receptor antagonists)
SELEXIPAG
Selexipag is an oral prostacyclin receptor agonist. It is given orally with initial dose of 200 μg 2 times a day. In the GRIPHON trial, selexipag relieved symptoms and delayed clinical deterioration.
RIOCIGUAT
Riociguat is a soluble guanylate cyclase stimulant and increases levels of cGMP levels which is a potent vasodilator. Riociguat is found to be effective in PAH and CTEPH patients. This drug should not be given in combination with phosphodiesterase factor 5 inhibitors (PDEF5) inhibitors.
SOTATERCEPT
PAH therapy is an evolving field and there are numerous clinical trials being done.New drugs and treatment modalities are being developed for the treatment of PAH in view of its progressive nature and bad prognosis in spite of treatment. Sotatercept is a first-in-class fusion protein that modulates TGF-β and BMPR2 signaling and is shown to have benefits in the recently completed Trial of sotatercept for treatment of pulmonary hypertension (STELLAR) trial. This may provide an additional fourth therapeutic pathway for the management of PAH. Additional therapies and routes of administration are under development, such as the inhaled tyrosine kinase inhibitor imatinib is being tried in clinical trials and did not show significant benefit.
PAH remains a progressive disease with a high mortality rate, despite the use of PAH drugs mainly targeting the imbalance of vasoactive factors.
Sotatercept is a fusion protein containing the extracellular domain of the human activin receptor type IIA linked to the Fc domain of human immunoglobulin G1—acts as a ligand trap for members of the TGF-β superfamily, thus restoring balance between growth-promoting and growth-inhibiting pathways. In phase 2 randomized controlled trial that included 106 patients with PAH treated over 24 weeks, subcutaneous sotatercept reduced PVR in patients receiving background PAH therapy; improvements were also observed in 6MWD and NT-proBNP. Stellar trial demonstrated significant benefit of sotatercept compared to placebo in patients with PAH who were receiving standard PAH medications with improvement in functional class, reduction of NT-proBNP levels, and PVR and it is approved by the States Food and Drug Administration for the treatment of PAH.[20]
HEART LUNG TRANSPLANTATION
Patients not responding to maximum medical therapy, patients with progressive PAH and estimated 1-year mortality more than 10%, and patients who are not going to respond to therapy like PVOD and capillary hemangiomatosis should be advised bilateral lung transplantation. Post-transplant median survival is 10 years in those who survive the 1st year. Heart– lung transplantation is advised in patients with Eisenmenger syndrome and complex congenital heart diseases.
BALLOON ATRIAL SEPTOSTOMY
Atrial septostomy may be considered as a palliative procedure or as a bridge to transplant in few patients with advanced PAH.[21] By unloading the failing right ventricle, this procedure results in shunt from the right to the left atrium, which may improve symptoms and hemodynamic parameters in advanced PAH. However, all patients are not suitable for this procedure because the shunt will cause oxygen saturation to decline.
PULMONARY ARTERY DENERVATION (PADN)
Several drugs targeting different pathophysiological mechanisms have become available over the last years for the treatment of PAH. Despite these drugs, long-term prognosis is poor with only 50% survival at 5 years.[22] Previous observational studies have suggested that hyperactivity of the sympathetic nervous system and the renin-angiotensin aldosterone system play a role in the pathophysiology of PH.[23,24] This data suggested that PADN may be a treatment option for these patients. The PADN procedure is considered experimental in the European Society of Cardiology guidelines for PH.[12]
Meta-analysis done by Salazar et al. showed that PADN improves hemodynamics with a significant reduction of PVR and m PAP with an increase in cardiac output. In patients with PH,[25] PADN therapy was also associated with improved functional capacity with a marked increase in 6MWD and better quality of life. Further randomized trials are required to demonstrate the procedure’s safety and efficacy for a long time.
SUPPORTIVE TREATMENT
General supportive measures play a major role in the management of patients with PAH. For patients with signs of hypoxia or volume overload, additional oxygen and diuretic therapies should be considered. All patients should receive vaccinations, psychiatric support, dietary guidance, and exercise regimens. In stable patients with PH, exercise training and rehabilitation are safe and effective measures in addition to medical therapy. Research indicates that these programs can enhance an individual with PAH’s ability to exercise, improve their hemodynamic profiles, their quality of life, and their overall well-being.[26] For women who are of reproductive age, pregnancy is contraindicated in view of high rate of maternal mortality in patients with PAH. Oral anticoagulation for idiopathic PAH, drug-associated PAH, or heritable HAP is not recommended in all patients as there is no evidence supporting the routine use of it. In these subgroups, decisions about empiric anticoagulation should be made on an individual basis.
CONCLUSION
Our understanding of PAH has significantly improved over the previous few decades. A thorough overview of the diagnosis and management of patients with PH is given in the ESC/ERS guidelines, fourth edition (2022). A revised hemodynamic definition of PH is mPAP >20 mmHg. This new definition’s therapeutic implications are extremely pertinent and of great clinical interest. In addition, suitable guidelines for PH centers have been suggested, and new algorithms for diagnosis, risk assessment, and treatment have been created. The significance of the TGF-β superfamily in the pathophysiology of the illness is now better understood, and several innovative treatments are being researched. Sotatercept has demonstrated advantages in improving functional class and NT-Pro BNP levels and PVR.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
Patient’s consent is not required as there are no patients in this study.
Conflicts of interest
Dr. Sridevi Chigullapalli is on the editorial board of the Journal.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Supplementary material:
Financial support and sponsorship
Nil.
References
- Primary Pulmonary Hypertension Geneva, Switzerland: World Health Organization; 1973. p. :15-7.
- [Google Scholar]
- An Overview of the 6th World Symposium on Pulmonary Hypertension. Eur Respir J. 2019;53:1802148.
- [CrossRef] [Google Scholar]
- The Prognostic Value of DLCO and Pulmonary Blood Flow in Patients with Pulmonary Hypertension. Pulm Circ. 2019;9:2045894019894531. doi:10.1177/2045894019894531
- [CrossRef] [Google Scholar]
- Genetics and Genomics of Pulmonary Arterial Hypertension. Eur Respir J. 2019;53:1801899.
- [CrossRef] [Google Scholar]
- Plexiform Lesions in Pulmonary Arterial Hypertension Composition, Architecture, and Microenvironment. Am J Pathol. 2011;179:167-79.
- [CrossRef] [Google Scholar]
- Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol Rev. 2019;99:1281-324.
- [CrossRef] [Google Scholar]
- New thoughts about the Origin of Plexiform Lesions. Am J Respir Crit Care Med. 2016;193:484-5.
- [CrossRef] [Google Scholar]
- The Role of Endothelin-1 in Pulmonary Arterial Hypertension. Glob Cardiol Sci Pract. 2014;2014:62-78.
- [CrossRef] [Google Scholar]
- Endothelin Receptor Antagonists in Pulmonary Arterial Hypertension. Eur Respir J. 2008;31:407-15.
- [CrossRef] [Google Scholar]
- Targeting Transforming Growth Factor-β Receptors in Pulmonary Hypertension. Eur Respir J. 2021;57:2002341.
- [CrossRef] [Google Scholar]
- 2022 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Eur Heart J. 2022;43:3618-731.
- [CrossRef] [Google Scholar]
- Die Sondierung des Rechten Herzens [Probing of the Right Heart] Klin Wochenschr. 1929;8:2085-7.
- [CrossRef] [Google Scholar]
- Complications of Right Heart Catheterization Procedures in Patients with Pulmonary Hypertension in Experienced Centers. J Am Coll Cardiol. 2006;48:2546-52.
- [CrossRef] [Google Scholar]
- Haemodynamic Definitions and Updated Clinical Classification of Pulmonary Hypertension. Eur Respir J. 2019;53:1801913.
- [CrossRef] [Google Scholar]
- Long-Term Response to Calcium Channel Blockers in Idiopathic Pulmonary Arterial Hypertension. Circulation. 2005;111:3105-11.
- [CrossRef] [Google Scholar]
- Predicting Survival in Pulmonary Arterial Hypertension: Insights from the REGISTRY to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010;122:164-72.
- [CrossRef] [Google Scholar]
- The REVEAL Registry Risk Score Calculator in Patients Newly Diagnosed with Pulmonary Arterial Hypertension. Chest. 2012;141:354-62.
- [CrossRef] [Google Scholar]
- Assessing Risk in Pulmonary Arterial Hypertension: What we know, what we don't. Eur Respir J. 2017;50:1701353.
- [CrossRef] [Google Scholar]
- Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. N Engl J Med. 2023;388:1478-90.
- [CrossRef] [Google Scholar]
- Effect of Atrial Septostomy on the Survival of Patients with Severe Pulmonary Arterial Hypertension. Eur Respir J. 2011;38:1343-8.
- [CrossRef] [Google Scholar]
- Pulmonary Arterial Hypertension: Pathogenesis and Clinical Management. BMJ. 2018;360:j5492.
- [CrossRef] [Google Scholar]
- Prognostic Significance of Sympathetic Nervous System Activation in Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2010;181:1269-75.
- [CrossRef] [Google Scholar]
- Potential Contribution of Carotid Body-Induced Sympathetic and Renin-Angiotensin System Overflow to Pulmonary Hypertension in Intermittent Hypoxia. Curr Hypertens Rep. 2019;21:89.
- [CrossRef] [Google Scholar]
- Pulmonary Artery Denervation as a New Therapeutic Option for Pulmonary Hypertension: A Systematic Review and Meta-Analysis. Curr Problems Cardiol. 2023;48:101776.
- [CrossRef] [Google Scholar]
- ERS Statement on Exercise Training and Rehabilitation in Patients with Severe Chronic Pulmonary Hypertension. Eur Respir J. 2019;53:1800332.
- [CrossRef] [Google Scholar]




