

Translate this page into:
Case Report of Rare Case of Cardiac Variant of Fabry’s Disease in a 2-Year-Old Child
Goutham Akidi, MD Department of Cardiology, Nizam’s Institute of Medical Sciences Panjagutta, Hyderabad 500082, Telangana India gouthamreddyakidi@gmail.com
This article was originally published by Thieme Medical and Scientific Publishers Private Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Fabry’s disease is caused by progressive lysosomal accumulation of neutral glycosphingolipids, primarily globotriaosylceramide. It results from deficiency of the enzyme α-galactosidase A, which is encoded by GLA on the X chromosome. Usually it presents in children as the classic variant, and the cardiac variant is extremely rare. Before labeling as cardiac variant in infants and children we should rule out other causes of the cardiac hypertrophy with left ventricular outflow tract obstruction like metabolic causes, hypertrophic cardiomyopathy in infants, Noonan’s syndrome, and nesidioblastosis.
We report a case of cardiac variant of Fabry’s disease in a 2-year-old male child. On evaluation he is found to have cardiac hypertrophy with no other features of Fabry’s disease, with low α galactosidase levels with no other systemic and syndromic features, which is an extremely rare presentation.
Keywords
Fabry’s disease
Alpha galactosidase
Hypertrophic cardiomyopathy
Noonan’s syndrome

Introduction
Fabry’s disease is caused by progressive lysosomal accumulation of neutral glycosphingolipids, primarily globotriaosylceramide; it results from the enzyme deficiency of α-galactosidase A, which is encoded by GLA on the X chromosome.1 As an X-linked condition, it mostly occurs in males with transmission by female carriers, although significant disease in later years can also be seen in women.2 3
The disease phenotype encompasses various signs and symptoms, with the following significant manifestations: angiokeratomas, acroparesthesias, anhidrosis, ocular changes, and eventually cardiovascular,4 cerebrovascular, and renal disease, all related mainly to the central pathophysiology of small-vessel vascular disease from the deposition of glycosphingolipid and consequent vascular insufficiency.
The hallmark of the classic early-onset disease in males in childhood usually causes severe pain in the extremities which is called as acroparesthesias, characterized by burning pain in the distal end of the extremities, triggered by a variety of stressors, and resulting in ischemia of peripheral nerves from the small-vessel disease. Angiokeratomas, red and purple punctate dermal lesions involving the lower midsection, buttocks, thighs, and upper part of the legs, may be one of the earliest signs of the disease. It accumulates progressively with age and age at onset, and phenotypic variability was related to the degree of α-galactosidase A deficiency, with less than 1% of the activity associated with the earliest and most aggressive disease2 3 (Table 1).
|
Manifestation |
Classic variant |
Variant of renal |
Variant of cardiac |
|---|---|---|---|
|
Onset age |
4–8 years |
More than 25 years |
More than 40 years |
|
Average death age |
41 years |
More than 60 years |
More than 60 years |
|
Angiokeratoma |
|
− |
− |
|
Acroparesthesias |
|
−/+ |
− |
|
Anhidrosis/hypohidrosis |
|
−/+ |
− |
|
Corneal/lenticular opacity |
|
− |
− |
|
Heart |
Left ventricular hypertrophy, ischemia |
Left ventricular hypertrophy |
Left ventricular hypertrophy, cardiomyopathy |
|
Brain |
Transient ischemic attack, stroke |
− |
− |
|
Kidney |
End stage renal disease |
End stage renal disease |
Proteinuria |
|
Residual α-galactosidase A enzyme activity |
Less than 1% |
More than 1% |
More than 1% |
Case Details
We report a case of a 2-year-old male child born out of nonconsanguineous marriage, fifth in the birth order, with the no significant similar complaints in other siblings. The eldest female sibling expired after two months of birth without any known congenital disease history, and the cause of death was not known.
This child was born after a full term of pregnancy at home and a history of a delayed cry for a 1 hour after birth. The birth weight was around 2.5 kg, and the child was hospitalized 20 days after birth with the features of lower respiratory tract infection (LRTI). During hospitalization, the child was diagnosed to have heart murmur but not in follow-up with cardiologist later. During the period of infancy there was a history of repeated hospital visits with the features of LRTI and history of delayed milestones during the growth period and poor weight gain and lack of growth. He was presented with this history to our institute and evaluated at the cardiology department.
On examination the weight of the child was 10 kg, subnormal to his age (2 years), and there were no facial abnormalities or any other dysmorphic features (Fig. 1). On cardiac examination, the child was found out to have a pan-systolic murmur at lower left sternal border.
-
Fig. 1 Facial features of the patient which is not suggestive of any syndromic appearance.
Fig. 1 Facial features of the patient which is not suggestive of any syndromic appearance.
Evaluated with the electrocardiography suggestive of sinus tachycardia with left axis deviation of around 90 to 100°, with right ventricular hypertrophy r>s, and t wave inversions in v1 and v2, equiphasic waves in v3,v4, and QS complex in v5 and v6.
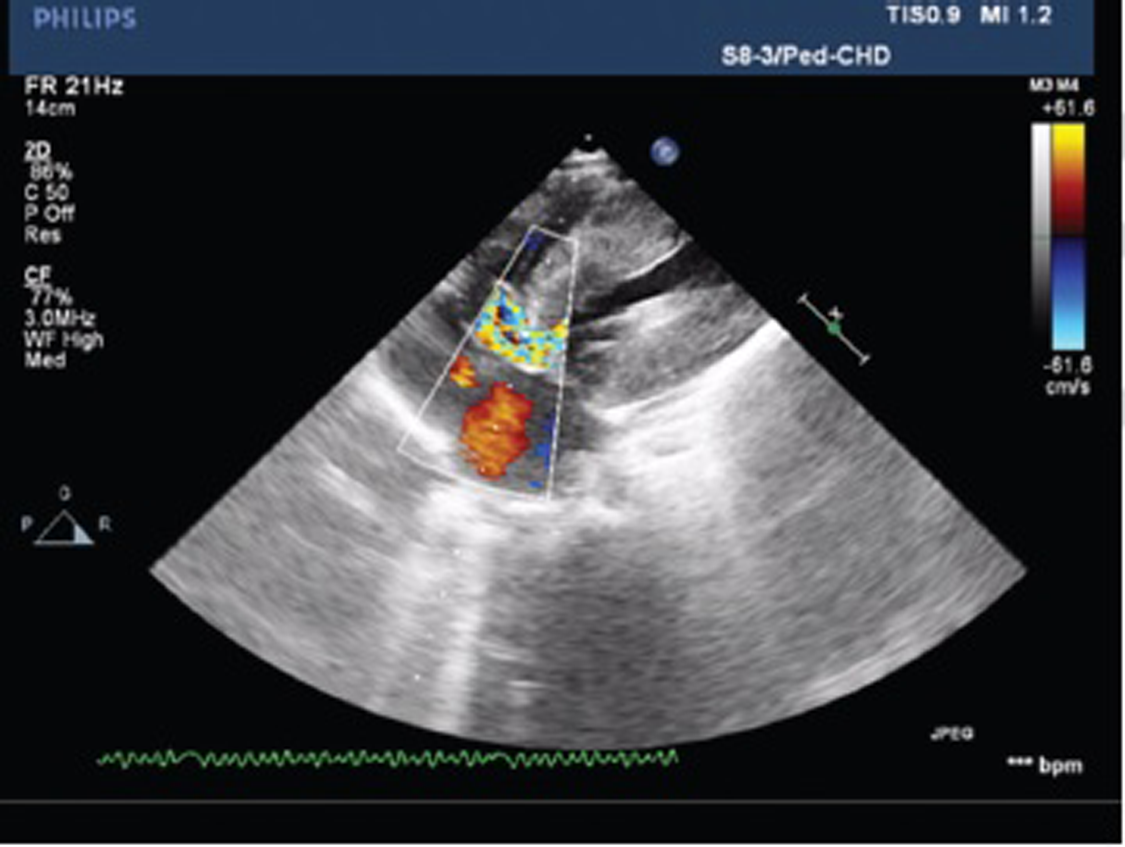
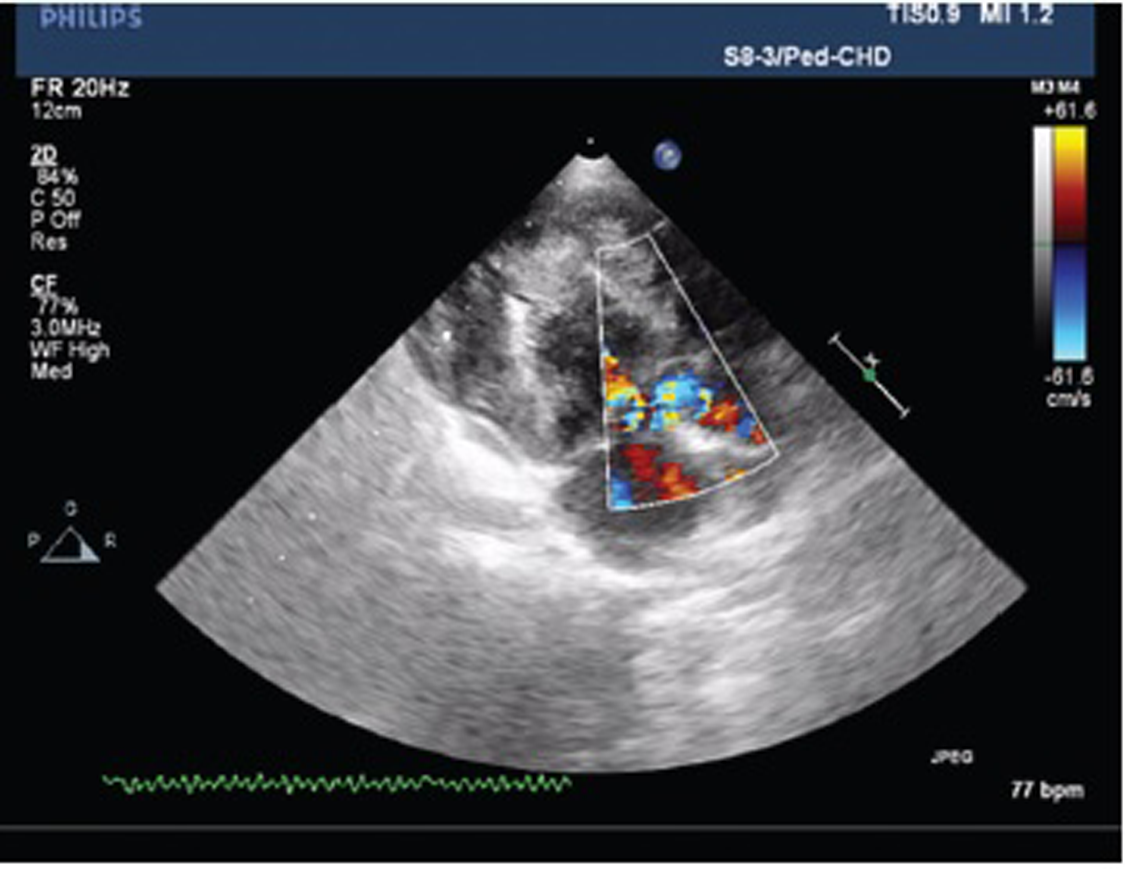
Echocardiography suggested that the patient had echo features of acyanotic congenital heart disease (ACHD) with moderately restrictive perimembranous ventricular septal defect (VSD) with the left to right shunt, concentric left ventricular hypertrophy (LVH), grade III systolic anterior motion of mitral valve (SAM), mild MR, resting left ventricular outflow tract (LVOT) gradient 50 mm Hg, and good biventricular function (Figs. 2 and 3).
-
Fig. 2 The apical five-chamber view of echo is showing cardiac muscle hypertrophy with left ventricular outflow tract obstruction.
Fig. 2 The apical five-chamber view of echo is showing cardiac muscle hypertrophy with left ventricular outflow tract obstruction.
-
Fig. 3 Plax view showing left ventricular outflow tract obstruction.
Fig. 3 Plax view showing left ventricular outflow tract obstruction.
Later the patient was evaluated with the α and β-galactosidase levels (nature of the specimen was blood leukocytes and the method was fluorometric assay using 4-methyl umbelliferone); β-galactosidase (117 out of 80–140) levels were in normal range with low levels ofα-galactosidase (03 out of 45–85 range). Planned for cardiac magnetic resonance imaging (MRI) but the patient was lost to follow-up.
Discussion
Physical examination is critical to detect certain causes of cardiac hypertrophy such as endocrine diseases like thyroid disease and acromegaly, LEOPARD and Noonan’s syndrome which are associated with specific facial dysmorphic features, and abnormal fat distribution like lipodystrophy. Metabolic diseases are usually associated with cataracts and neuromuscular problems. Signs of ataxia are present in syndrome like Fredrich’s ataxia. So it is essential to look for physical examination and various facial features and clinical signs for certain syndromes. The syndromes discussed earlier are usually associated with the cardiac hypertrophy; so it is important to rule out these diseases in infants and children.
Preliminary metabolic screening tests to diagnose certain causes of hypertrophic cardiomyopathy should include fasting blood sugar, lactate, carnitine, pyruvate, organic acids, amino acids and urine amino acids, vacuolated lymphocytes, and glycosaminoglycans.
In infants and children, this is the various causes of cardiac hypertrophy.
Metabolic Causes of Cardiac Hypertrophy
Metabolic disorders which are associated with the hypertrophic cardiomyopathy manifest in early childhood. Typically, the cardiomyopathy in metabolic syndromes are either dilated or hypertrophic and is usually associated with a decreased systolic function. Specific conditions include fatty acid metabolism; some of them may respond to carnitine. Usually, they are autosomal recessive abnormalities.5 In metabolic diseases, infection or fasting of children may lead to cardiac failure and even sudden cardiac death.
Disorders of mitochondrial oxidative phosphorylation usually exhibit maternal inheritance are usually associated with neuromuscular disease and can present in childhood or adult life.6 7
Myocardial infiltration with amyloid and sarcoid can result in cardiac hypertrophy in all age groups but common in adult life and rare in infancy or childhood. Glycogen storage diseases are more common causes of cardiomyopathy, predominantly types II and III. Type II is usually lethal in infancy and the prenatal diagnostic tests available for this disease.8 Other diseases in which metabolites accumulate in the myocardium and lead to hypertrophy of myocardium are Gaucher’s disease, GM1 and GM2 gangliosidosis, sialidosis, and mannosidosis.
Syndromes Associated with Cardiac Hypertrophy
Noonan’s syndrome is one of the most common causes of childhood cardiac hypertrophy; one-third cases of childhood hypertrophy occur because of Noonan’s syndrome. LEOPARD syndrome is a variant of Noonan’s syndrome; both are autosomal dominant diseases associated with the features like multiple lentigines or café au lait spots, ocular hypertelorism, pulmonary valve stenosis, and cardiac hypertrophy. These disorders associated with the mutations in RAS signaling. So, they called as RASopathys. Evaluation for RASopathys is essential in childhood hypertrophic cardiomyopathys.
Histological picture of the myocardium of this syndrome is similar to hypertrophic cardiomyopathy of familial type,9 but echo features will show minimal differences. 25% of patients with Noonan’s syndrome will have cardiomyopathy,10 but it is unusual to have sudden cardiac death. Noonan’s syndrome with cardiac hypertrophy in adults is sporadic compared with childhood because mortality is more common in childhood with heart failure. They rarely survive beyond childhood and facial features vary with age in adulthood in those who survive beyond childhood; so it is challenging to recognize morphologically in adulthood.11
Gestational Diabetes and Nesidioblastosis
In infants who are born with the hypertrophic cardiomyopathy, it is essential to rule out the presence of gestational diabetes12 because gestational diabetes is one of the causes of cardiac hypertrophy in newborns and infants. Nesidioblastosis can be associated with the cardiac hypertrophy; so it should be consider as one of the differential diagnoses in infant and childhood cardiac hypertrophy. In nesidioblastosis hypertrophy is reversible once hyper insulin levels are controlled by management of the disease.
Familial Hypertrophic Cardiomyopathy
It is inherited as an autosomal dominant pattern in 50% of cases; in remaining cases, it will detect as de novo without any family history.
Fabry’s Disease with Cardiac Hypertrophy in Childhood
Fabry’s disease is a storage disorder it inherited as X-linked pattern, with defective lysosomal enzyme α-galactosidase A activity. Usually, the cardiac variant of Fabry’s disease prevalent in the age group older than 40 years. It is extremely rare in childhood. In this case the patient has hypertrophic cardiomyopathy with low α-galactosidase activity levels suggesting a variant of Fabry’s disease because there are no other features of Fabry’s disease that are manifested in this case and no other abnormalities suggesting metabolic or syndromic causes of cardiac hypertrophy.
One newborn screening study in Taiwan Chinese population, conducted by Lin et al13 shows an increased incidence of Fabry’s disease of the cardiac variant in which 16 males who had been diagnosed with idiopathic hypertrophic cardiomyopathy were screened by mutation analysis and α-galactosidase activity in combination with intronic mutation showed deficient enzyme levels and mutations in 4 cases (25%); this study shows IVS4+919G→A14 mutation among both newborns (1 in 1,600) and patients with idiopathic cardiomyopathy.
Treatment options available are enzyme replacement therapy (ERT) that supplies recombinant enzyme to cells which are deficient in it, and it reverses the pathologic and metabolic abnormalities. It is available for Fabry’s disease since 2001, and it is to be given second weekly.15 16 Available recombinant preparations are of two types—alfa GLA (Replagel) to be given 0.2 mg per kg infusion, and β GLA preparation (Fabrazyme) which is to be given 1 mg per kg infusion; only β GLA preparation is FDA approved. Other therapies which are available for Fabry’s disease are substrate reduction therapy, residual enzyme activation, GLA promotor activation, protein homeostasis regulation (proteostasis), chemical chaperon therapy, and small molecular compounds which can target various cellular structures and functions and by that it can alter cellular function.
Conclusion
In this case report, the patient had retarded growth and recurrent LRTIs, and echo features represent LVOT obstruction. No other systemic or syndromic features were present. We evaluated for Fabry’s disease, which is one of the rare diseases in young children, and the cardiac variant is extremely rare. Usually, a classic variant will manifest in early childhood, but this child had a cardiac variant of Fabry’s disease, and enzyme assay showed lower levels of α-galactosidase which is a defective enzyme in Fabry’s disease.
In infants or young children, we should evaluate for these causes of cardiomyopathy before labeling as idiopathic.
Conflict of Interest
None.
References
- Fabry disease. In: Pagon RA, Adam MP, Ardinger HH, et al. eds. Gene Reviews. Seattle, WA: University of Washington: Seattle 2002 [Initial posting August 5; last update January 5
- Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007;30(02):184-192.
- [Google Scholar]
- Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008;93(02):112-128.
- [Google Scholar]
- Cardiac abnormalities in Anderson-Fabry disease and Fabry’s cardiomyopathy. Cardiovasc J Afr. 2011;22(01):38-44.
- [Google Scholar]
- Impaired skin fibroblast carnitine uptake in primary systemic carnitine deficiency manifested by childhood carnitine-responsive cardiomyopathy. Pediatr Res. 1990;28(03):247-255.
- [Google Scholar]
- Prenatal diagnosis of Pompe’s disease (type II glycogenosis) in chorionic villus biopsy using maltose as a substrate. Prenat Diagn. 1992;12(03):169-173.
- [Google Scholar]
- Cardiologic abnormalities in Noonan syndrome: phenotypic diagnosis and echocardiographic assessment of 118 patients. J Am Coll Cardiol. 1993;22(04):1189-1192.
- [Google Scholar]
- Characterization of the cardiomyopathy in infants of diabetic mothers. Circulation. 1980;61(02):441-446.
- [Google Scholar]
- High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ Cardiovasc Genet. 2009;2(05):450-456.
- [Google Scholar]
- Novel α-galactosidase A mutation in patients with severe cardiac manifestations of Fabry disease. Gene. 2014;535(02):365-369.
- [Google Scholar]
- Clinical results of enzyme replacement therapy in Fabry disease: a comprehensive review of literature. Int J Clin Pract. 2007;61(02):293-302.
- [Google Scholar]




