

Translate this page into:
ABCC9 Associated Symptomatic Advanced Atrioventricular Block in a Patient with Significant Family History of Sudden Cardiac Death: A Case Report
*Corresponding author: Aditya Sharma, Department of Cardiology, Nizam Institute of Medical Sciences, Hyderabad, Telangana, India. dradityasharma0310@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Maddury J, Sharma A, Imran S. ABCC9 associated symptomatic advanced atrioventricular block in a patient with significant family history of sudden cardiac death: A case report. Indian J Cardiovasc Dis Women 2022;7:210-3.
Abstract
Sudden cardiac arrest (SCA) and its consequence sudden cardiac death (SCD) are the common cardiac pathway for death. Often, the cause is not found and the inability to delineate the underlying process presents a major public health challenge. Although CAD represents the most common cause of SCD, arrhythmias are an important cause of the same as these patients present with fewer premonitory symptoms and often go undetected. Inherited arrhythmia syndromes represent a challenge due to limited availability of widespread genetic testing and known pathogenic genetic mutations. One such gene is ABCC9 gene, which encodes the SUR2 subunit of the ATP sensitive potassium channel or KATP channel. Mutations in KATP channel are associated with wide range of inherited diseases. Gain-of-function mutations are associated with Cantu syndrome characterized by hypertrichosis and acromegaloid facial features. Loss-of-function mutations are associated with Brugada syndrome and dilated cardiomyopathy. Here, we report a patient with a likely pathogenic mutation in the ABCC9 gene, identified by whole exome sequencing. The male proband came with multiple episodes of syncopal events and palpitations found to have advanced atrioventricular block. This case highlights the consequences of KATP channel dysfunction in the cardiovascular system.
Keywords
ABCC9
Sudden cardiac death
Atrioventricular block
Bradyarrhythmia

INTRODUCTION
The ABCC9 gene encodes a transmembrane protein SUR2 that forms the regulatory part of the ATP-sensitive potassium channel (KATP). These are majorly located in cardiac, skeletal, and vascular smooth muscle cells.[1] KATP channels are hetero-octamers consisting of four pore-forming Kir6.x subunits and four regulatory SUR (sulfonylurea receptor) subunits. The pore forming units are produced from either the KCNJ8 or KCNJ11 gene, whereas the regulatory SUR units are produced from the ABCC8 gene (SUR1) or ABCC9 gene (SUR2). The SUR2 subunits are responsible for regulating the activity of the KATP channels. These channels adapt the potassium ion conductance off cell membrane in response to the ATP content of the cell.
Here, we present a case of a likely pathogenic ABCC9 mutation in a patient presenting with advanced atrioventricular (AV) block.
CASE REPORT
The patient, a 39-year-old male, driver by occupation, presented with history of syncopal events: Four episodes over the preceding 1 week before admission. Syncope episodes were not related to exertion and were preceded by giddiness, shortness of breath, and palpitations. The patient never had chest pain, orthopnea, and paroxysmal nocturnal dyspnea. There was no history of similar episodes in the remote past. The patient had no other comorbidities. He denied any addictions. On enquiry, similar episodes of syncope were present in his sister and father [Figure 1]. Patient’s father had pacemaker implanted at the age of 46 years after which he discontinued follow-up in hospital and died at the age of 56 years due to sudden cardiac arrest. Patient’s sister had pacemaker inserted at the age of 35 years due to symptomatic bradycardia. Medical records of the family were not available.
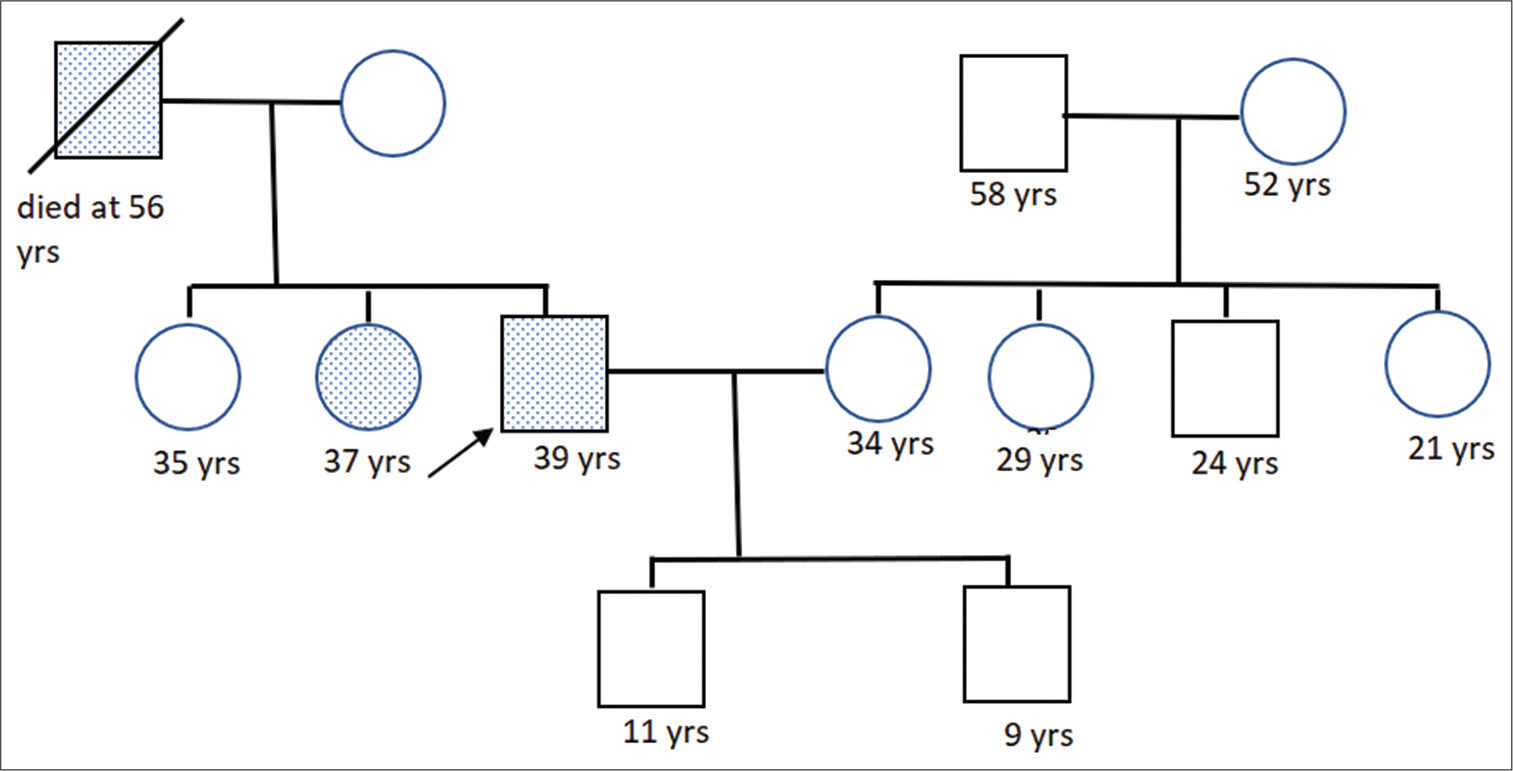
- Pedigree chart for the proband.
On admission, the patient was vitally stable with a regular pulse of 80/min. Cardiovascular examination was unremarkable. The patient’s blood investigations, including complete blood panel, renal parameters including electrolytes, liver function, and thyroid function tests, were all unremarkable. Hs-Trop I was also within normal limits. Resting ECG was suggestive of complete left bundle branch block with first degree AV block with a PR interval of 240 ms and corrected QT of 468 ms [Figure 2]. During observation in ICU, the patient also developed intermittent complete heart block. In view of episodic syncope, 24-h Holter was attached and was suggestive of few episodes of Mobitz Type 1 AV block and advanced AV block during which time the patient had giddiness [Figure 3]. A 2D echocardiography was suggestive of no regional wall motion abnormality, normal chamber sizes, left ventricular dimensions of 42 × 31 mm with a LVPW and IVS thickness of 9 mm each, and good biventricular systolic function with grade 2 LV diastolic dysfunction. Invasive coronary angiogram done was suggestive of normal epicardial coronaries.
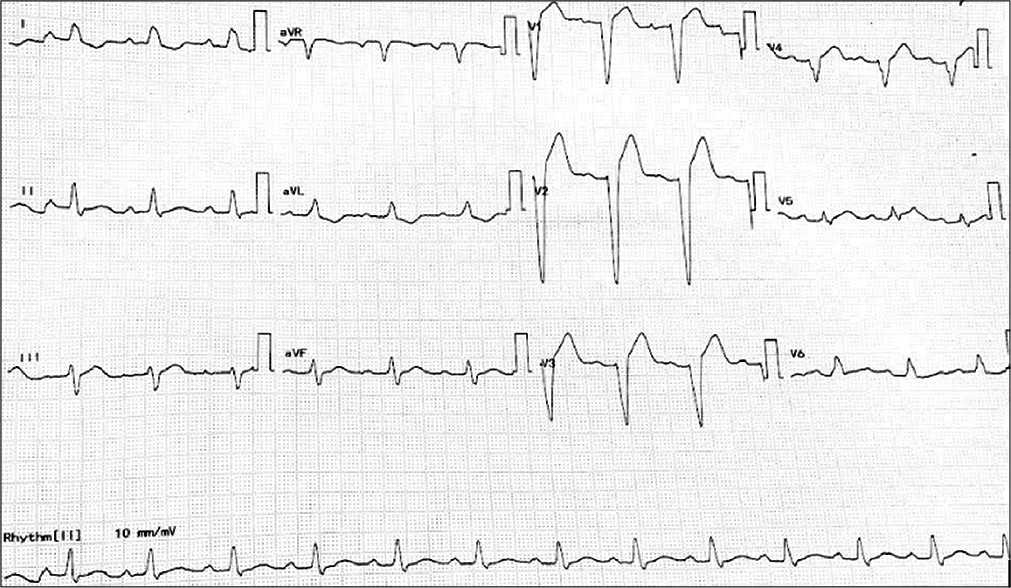
- Standardized 12 lead ECG of the proband.
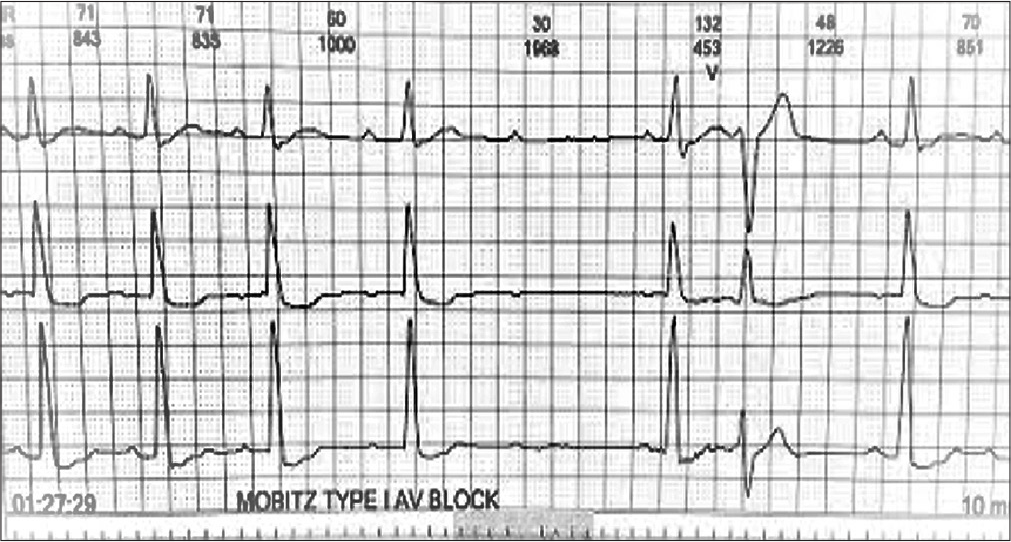
- Holter recording showing Mobitz type 1 second degree Atrioventricular Block.
Cardiac MRI was done which revealed normal chamber size and ejection fraction, without any late gadolinium enhancement, and no findings suggestive of myocarditis or arrhythmogenic ventricular dysplasia. In view of significant family history, next-generation sequencing for monogenic causes of arrhythmia (whole exome sequencing panel) was done to see for inherited arrhythmia syndromes. All disease-related genetic variants were classified according to the American College of Medical Genetics (ACMG) guidelines which showed ABCC9 mutation positive. ECG and 2D echo for his sisters and mother were also advised.
Whole exome sequencing was suggestive of a heterozygous mutation in the ABCC9 gene in intron 3 location: c.142+5G>A; 5’ splice variant. Variant was classified as pathogenic as per ACMG guidelines and as disease causing by mutation prediction software MutationTaster2. As per databases (1000 genomes and internal laboratory database), this mutation has not been previously reported in other patients and has a minor allelic frequency of 0.0007% in gnomAD database.
As this mutation is rare among the population, the risk of ventricular arrhythmias is not reported in the literature. Due to the lack of available literature and the fact that mutations in this gene are associated with development of dilated cardiomyopathy and possible future risk of ventricular tachycardia/fibrillation, decision was taken for dual chamber automatic implantable cardioverter-defibrillator (AICD) implantation. Post-procedure the patient was monitored for 72 h during which he had no further symptoms. AICD interrogation at 1 week showed <1% of atrial and ventricular paced beats with no shocks delivered.
DISCUSSION
ATP sensitive potassium channel or KATP channels are primarily localized in cardiac, skeletal, and vascular smooth muscle cells. It is a type of potassium channel that is gated by intracellular nucleotides, namely, ATP and ADP. It is a hetero-octamer consisting of eight subunits. As they couple the metabolic state of cell to its membrane potential, changes in the cellular metabolites such as glucose or fatty acids can alter the activity of these channels. Therefore, dysfunction in the KATP channel leads to the abnormal response to metabolic stress.
The gain-of-function or loss-of-function mutations of various genes encoding KATP subunits are associated with different phenotypes. Regulatory subunit encoded by ABCC8 and the pore-forming subunit encoded by KCNJ11 are expressed in insulin-secreting tissues. Gain-of-function mutations in these subunits are associated with neonatal diabetes, whereas loss-of-function mutation is associated with hyperinsulinism.[2] However, mutations in SUR1-and ABCC8 are not reported to be associated with cardiovascular phenotypes.
ABCC9 gain-of-function mutations are associated with Cantu syndrome, which presents with hypertrichosis, acromegaloid facial features, and cardiomegaly.[3] ABCC9 loss of function mutations have been reported only twice, once in a case presenting with dilated cardiomyopathy and the second time in a case presenting with paroxysmal atrial fibrillation.[4,5]
Here, we report a patient with a likely pathogenic variant in the ABCC9 gene associated with a unique phenotype, that is, advanced AV block. In addition, we did not find any evidence of adverse cardiac remodeling based on echocardiography and cardiac MRI. However, it may be associated with development of dilated cardiomyopathy and subsequent arrhythmias later, as has been associated with this genotype, hence the decision to go for a dual chamber AICD over a dual chamber pacemaker without defibrillation function. The case described identifies ABCC9 gene mutation as a likely disease-causing candidate for inherited arrhythmia syndrome. It also highlights the importance of the use of wider target genetic panels in patients presenting with arrhythmias without any evidence of structural heart disease.
Several limitations must be mentioned here. First is the lack of functional studies for the variant described and the inability to make a strong conclusion regarding the impact of the variant on the phenotype. Second, the genotypic determination of the phenotype positive as well as phenotype negative family members remains to be determined to assess if the variant found can be a likely polymorphism rather than a pathogenic variant. Third, long-term follow-up and assessment of the proband’s clinical status and device interrogation are awaited.
CONCLUSION
We presented a case of a heterozygous mutation in ABCC9 gene in a case presenting with advanced AV block without any evidence of structural heart disease. This case, further, adds understanding to the knowledge of disorders associated with mutation in ABCC9 gene and sheds light on the consequences of KATP dysfunction. It adds to the phenotypic presentation of ABCC9 mutation associated diseases. It also underscores the importance of KATP channels in cardiomyocyte electrophysiology, function, and their arrhythmogenic potential.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Cloning, tissue expression, and chromosomal localization of SUR2, the putative drug-binding subunit of cardiac, skeletal muscle, and vascular KATP channels. Diabetes. 1996;45:1439-45.
- [CrossRef] [PubMed] [Google Scholar]
- The genetic and molecular mechanisms of congenital hyperinsulinism. Front Endocrinol. 2019;10:111.
- [CrossRef] [PubMed] [Google Scholar]
- Cantú syndrome: Findings from 74 patients in the international cantú syndrome registry. Am J Med Genet C Semin Med Genet. 2019;181:658-81.
- [CrossRef] [PubMed] [Google Scholar]
- ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004;36:382-7.
- [CrossRef] [PubMed] [Google Scholar]
- KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat Clin Pract Cardiovasc Med. 2007;4:110-6.
- [CrossRef] [PubMed] [Google Scholar]




